



Have a Thick Differential Diagnosis List for Thick Hearts
To Avoid Missing A Treatable Disease

Reproduced by Shutterstock by Subscription
“Diagnostic anchoring” refers to clinicians’ tendency to make a diagnosis and to avoid considering the alternatives. I have had several patients referred to me with the diagnosis of hypertrophic cardiomyopathy (HCM), but who turned out to have other disease that can produce increased left ventricular wall thickness. These conditions are sometimes called “HCM phenocopies” because they copy the HCM phenotype but do not have the genotype. Notice that I said “increased left ventricular wall thickness” and not “left ventricular hypertrophy” (LVH). LVH really means cardiac muscle hypertrophy and not just a thick wall.
Recognizing HCM phenocopies, and not succumbing to diagnostic anchoring, is critically important because there are now treatments for some HCM phenocopies that can prevent progression and even initiate regression of the non-HCM disease. Let’s review the differential diagnosis.
The differential diagnosis of a thick heart includes classical HCM, long-standing poorly treated hypertension, myxedema heart from hypothyroidism, cardiac amyloidosis, alpha galactosidase A (a-GSA) deficiency or Fabry disease, lysosomal associated membrane protein–2 deficiency (LAMP-2) disease also called Danon disease, and hydroxychloroquine (HCQ) treatment, which President Trump previously suggested as COVID-19 therapy.
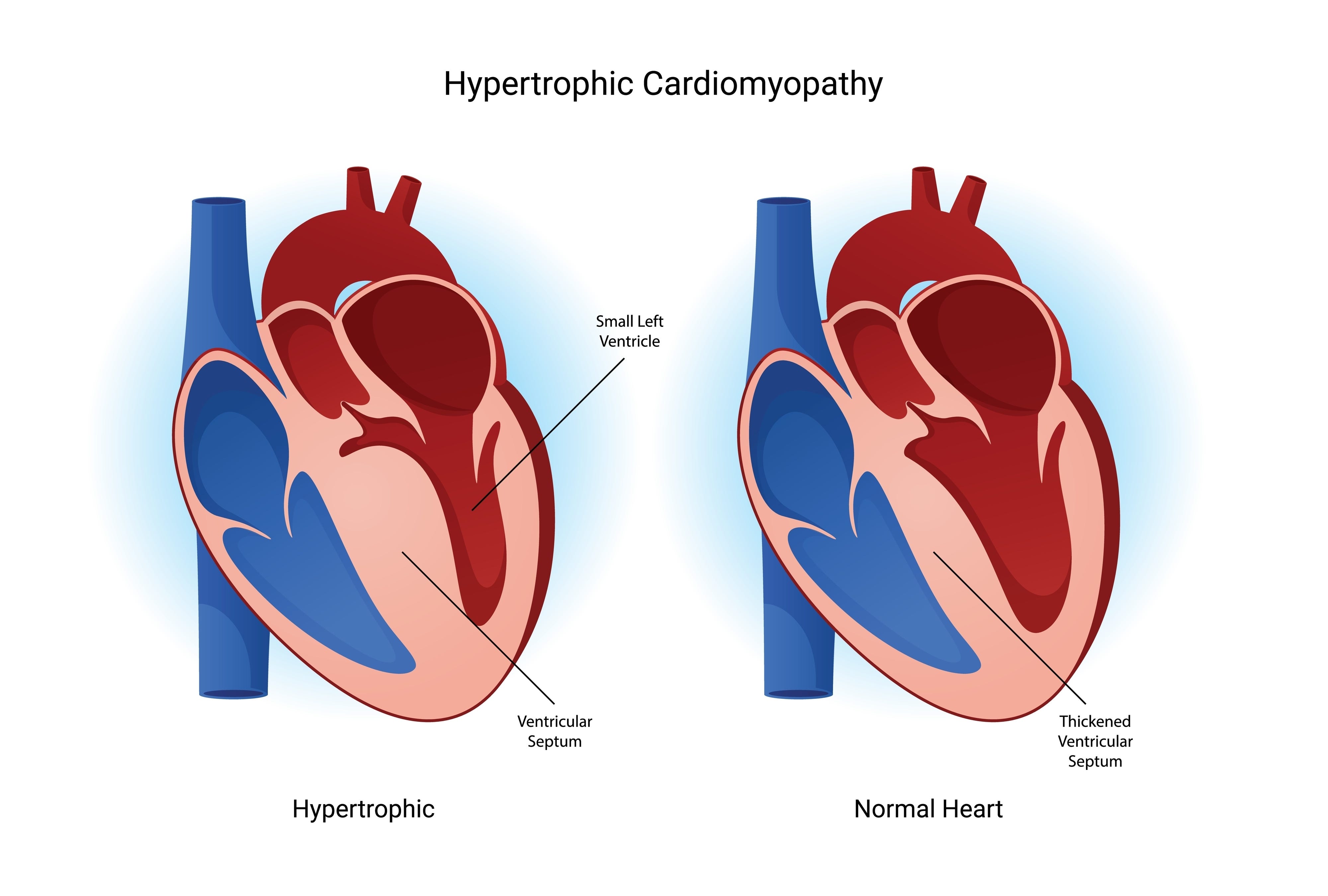
HCM is the most frequent cause of thick hearts and affects 1/250 individuals. HCM is autosomal dominant and not sex linked. It typically, but not always, produces more septal than other wall hypertrophy. Indeed, HCM was historically called asymmetric septal hypertrophy or ASH. Clinicians should consider diagnoses other than HCM if there is no family history of HCM, when the disease onset is late in life, and when there is symmetric LVH or LVH that does not include the septum. But be careful because if anything typifies HCM, it is its variability. HCM can produce apical only and symmetrical LVH, appear late in life, and occur in non-inherited patterns. My friend, Dr. Barry Maron, a pioneer researcher in HCM, has also reported a “non-inherited” cluster of HCM patients that was due to sperm-donation from an undiagnosed HCM carrier.(1)
Long-standing, poorly treated hypertension (HBP) can cause LVH, as can marked hypothyroidism, and the wall thickness can be quite remarkable in both cases. On my last in-patient service stint, we consulted on a man with long-standing hypertension and heart failure with preserved ejection fraction whose left ventricular septum and posterior wall were 18 and 17 mm in thickness, respectively. Normal wall thickness is 1o mm or less.
Cardiac amyloidosis is presently recognized more frequently as the cause of thick hearts because of increased awareness of the disease and the use of non-echo imaging techniques such as magnetic resonance imaging (cMRI) and technetium 99 pyrophosphate scanning. Amyloidosis comes in three varieties. (2) Deposition of antibody light chains produces AL amyloidosis. These light chains are produced by clonal plasma cells so it is treated as a variant of multiple myeloma. Inherited transthyretin (TTR) amyloidosis is due to mutations in the TTR gene. A gene mutation is present in approximately 3% of African Americans, but mutations occur in other populations including the Irish. There is also a non-inherited or “wild type” (wt) TTR amyloidosis. wt TTR amyloidosis is more frequent in men, especially old men, despite not being sex-linked. All types of amyloidosis produce orthopedic issues such as biceps tendon ruptures, carpal tunnel symptoms, trigger fingers, palmar tendon nodules and spinal stenosis. These orthopedic issues may appear years before the cardiac amyloidosis becomes obvious. This site has discussed amyloidosis. (3)
Amyloidosis should also be considered in patients with LVH and peripheral neuropathy since amyloid also invades peripheral nerves. It is important to diagnose TTR amyloidosis because new treatments such as tafamidis (4) and coramidis (5) can preserve functional capacity and reduce cardiovascular mortality. Interestingly, 20% of patients with wtTTR have asymmetric septal hypertrophy making wtTTR a worthy HCM phenocopy. (3)
Patients whose LVH is due to HCM or high blood pressure (HBP) should have impressive electrocardiographic voltage criteria for LVH whereas patients with amyloidosis have either normal or decreased LV voltage. A history of HBP that has been mysteriously cured and no longer requires anti-HBP drugs suggests new peripheral neuropathy another symptom of amyloidosis. The neuropathy reduces arterial vasoconstriction reducing the blood pressure.
Clinicians should also think of amyloidosis in any patient with orthopedic or tendon issues. I have diagnosed cardiac amyloidosis in a surgeon with biceps tendon rupture, a geneticist with three trigger finger surgeries, and an ophthalmologist with bilateral carpal tunnel surgery. I only pursued the diagnosis because of their tendon issues. A normal echo does not exclude the disease because approximately 10% of patients with cardiac amyloidosis have normal left ventricular wall thickness.
Fabry disease is another treatable cause of thick hearts. Fabry is X-linked and results from a decrease in lysosomal alpha galactosidase A (a-GSA) activity resulting in the accumulation of glycosphingolipids in lysosomes. About 0.03% of the population carries the Fabry gene. It usually produces concentric LVH with late gadolinium enhancement (LGE) of the posterior wall and, in contrast to HCM, spares the septum.
Fabry also can produce ascending aortic enlargement and aortic insufficiency. Fabry should be considered with late onset left ventricular wall thickening especially if there is aortic enlargement. The disease is more common and severe in men, because of the X-linked inheritance. This inheritance pattern means that men can be diagnosed by measuring serum a-GSA activity because they don’t produce any, but that women must have genetic analysis to detect the genetic defect. Fabry also causes AV conduction disease. Even though Fabry usually causes diffuse LVH, it can produce septal only hypertrophy and even systolic anterior motion of the mitral valve so Fabry should be excluded in all thick hearts. Both enzyme replacement therapy and migalastat, which helps chaperone a-GSA to lysosomes, reduce LVH in Fabry.(6)
Danon or LAMP-2 disease is another rare, X-linked lysosomal glycogen storage disease. It usually presents in children who also have muscle weakness and intellectual disability.
Hydroxychloroquine (HCQ) can also produce a lysosomal storage disorder that mimics HCM because HCQ inhibits lysosomal function.(7) HCQ is not recommended for COVID-19 treatment but is still used to treat rheumatoid arthritis and other inflammatory diseases, so should be considered as a cause of LVH in patients treated with this drug.
We are all taught to make a differential diagnosis for every patient’s condition, but this is especially important with thick-hearted patients to avoid missing a treatable disease.
1. Maron BJ, Lesser JR, Schiller NB, Harris KM, Brown C, Rehm HL. Implications of hypertrophic cardiomyopathy transmitted by sperm donation. JAMA. 2009;302(15):1681–1684.
2. https://substack.com/home/post/p-157352155
3. Ruberg FL, Grogan M, Hanna M, Kelly JW, Maurer MS. Transthyretin amyloid cardiomyopathy: JACC state-of-the-art review. J Am Coll Cardiol. 2019;73(22):2872–2891.
4. Maurer MS, Schwartz JH, Gundapaneni B, et al. Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med. 2018;379(11):1007–1016.
5. Gillmore JD, Judge DP, Cappelli F, Fontana M, Garcia-Pavia P, Gibbs S, Grogan M, Hanna M, Hoffman J, Masri A, Maurer MS, Nativi-Nicolau J, Obici L, Poulsen SH, Rockhold F, Shah KB, Soman P, Garg J, Chiswell K, Xu H, Cao X, Lystig T, Sinha U, Fox JC. Efficacy and Safety of Acoramidis in Transthyretin Amyloid Cardiomyopathy.; ATTRibute-CM Investigators.N Engl J Med. 2024 Jan 11;390(2):132-142.
6. Germain DP, Hughes DA, Nicholls K, et al. Treatment of Fabry disease with the pharmacologic chaperone migalastat. N Engl J Med. 2016;375(6):545–555.
7. Yogasundaram H, Putko BN, Tien J, et al. Hydroxychloroquine-induced cardiomyopathy: Case report, pathophysiology, diagnosis, and treatment. Can J Cardiol. 2014;30(12):1706–1715.
#thickhearts #cardiachypertrophy #hypertrophiccardiomyopathy #hypertension #Fabrydisease #LAMP #hydroxychloroquine #tafamidis
So easy to read and a bottom up approach to LVH. Wonderful writing on a somewhat difficult differential diagnosis. May I suggest you extend your talents to Pulmonary Hypertension and also write a comprehensive commonsense review? Thanks again..
Thank you for the kind comments. I will consider doing something on pulmonary hypertension, but not soon since I don't consider myself expert enough. Paul